序言
本系列是我对三年来学习Linux,R,Python,从寻找在线服务器到自己写在线服务器的过程的回忆,记录与总结,不会介绍太多生物学的背景知识,适合于对生物学有了解的读者。内容涉及:
- 基因组注释/功能基因组学
- 转录组/芯片数据分析
- 蛋白质结构预测,分子对接
- 基于结构/序列信息的蛋白质性质改造/预测
- 简单的Python、Linux和R的介绍
我关注了不少计算和生信的教学和论坛,但是我觉得许多都偏向医学领域。目前计算生物学和生物信息学应用于极端微生物的研究,许多性能独特的酶被发现或被改造,感觉需要向工业生物技术领域的研究人员介绍计算生物学和生物信息学的使用。整个系列不侧重理论(生物学基础和软件的算法都不侧重),重点在于介绍这些方法和讨论这些方法如何应用于指导湿实验。
在此我想要感谢明灯明研究员对我的指导、帮助和关心。正是因为他的耐心培养使得这个系列有了出现的可能。
第一章 LINUX基础
Linux与Ubuntu
还记得黑客们没有图标的屏幕上跳动的绿色字符么,而且他们似乎完全不需要鼠标。

其实在出现可视化界面之前,搞计算机的都在终端里面敲命令行。对于生物信息学相关的软件来说,基本上只有在线服务器和一些昂贵的收费软件是通过点击鼠标完成的,大部分的软件还是需要在终端里通过命令行运行的,平时的文件格式的处理什么的工作也会在命令行里得到快速的完成。
Windows有CMD,Mac有自己的Treminal,但是我们要向大家介绍一个新的系统:Linux。
Linux是一类开源的操作系统,在服务器上基本上都运行着Linux,Ubuntu属于其中的一个发行版,也是整个教程依赖的操作系统。
伴随着云端技术的发展,我们每个人都可以拥有一台自己的云服务器。建议读者注册一个阿里云或者腾讯云,学生套餐一年也就一百多,具体怎么注册我就不赘述了,选择系统镜像时选择Ubuntu。
重要的事情说三遍:Linux,Linux,Linux!
Python基础与实例
Python(念作:‘拍僧’,我曾经有个同学和我说他他在学 ‘非统’ 😂),可以说是这个时代最流行的语言了,风格及其接近伪代码,现在很多大学理工科专业的程序语言设计课程都从VB、C++换成了Python。Python 2.x 版本在2020年元旦停止了支持(21世纪20年代第一件大事啊)。这里就不再回顾发展与转型的阵痛了,总之我们可以肯定Python 3.x 作为新事物已经战胜了旧事物。
Python之所以受欢迎一方面是简单代码少,可读性强(缩进和空格非常严格),更是因为强大的开源社区提供了数以万计包package只需要简单的import就可以调用许多功能强大的函数。所谓函数其实就是A->B,我们输入A,得到B。
这一份教程不会从变量开始一点点介绍Python,我曾经系统学习过,但是很快就忘光了,还是在解决问题的过程中学习的比较扎实,我会通过一个个实用的例子讲解Python的具体用法。我们会用到它处理文件,做简单的计算,写一些简单的流程,也会涉及一些机器学习。
2.1 使用Python处理FASTA文件
FASTA格式的序列文件以单行描述开始,然后是序列数据行。 描述行(defline)用大于号>开头与序列数据行区分开来。 FASTA格式的示例序列为:
>P01013 GENE X PROTEIN (OVALBUMIN-RELATED) QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS VLMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRADHP FLFLIKHNPTNTIVYFGRYWSP
下面我们通过介绍处理FASTA文件来介绍Python的基本用法。
2.1.1 提取FASTA
问题:我们有一个含有几十万条蛋白序列的巨大的FASTA文件,现在要从中分离出给定的序列(好希望有一根智能柱子)。
方法:把FASTA文件做成序列名称和序列对应的结构。想象成抗体和抗原。
字符串是Python中的一种数据类型,一切都可以被看作字符串,字符串不可变,就是符号串在一起。
字典是Python中一种可变容器模型,且可存储任意类型对象。
字典的每个键值对( key=>value) 用冒号:分割,每个键值对之间用逗号 , 分割,整个字典包括在花括号 {} 中 ,格式如下所示:
d = {key1 : value1, key2 : value2 }
其中键是不可以重复的,对同一个键多次赋予不同的值,只会保留最后一个。具体可参见字典。
sys是一个python内置的包,用于终端的交互。
#开头的行在Python代码中是注释,解释器不会运行这一行,只是给你看的。
import sys
#我们使用 def functionname(): 定义一个函数,方便调用。
def fasta_2_dic(fasta):
#创建一个名叫dic的空字典,我们将要建立序列名称和序列的一一对应关系。
dic = {}
#fasta是被传入函数的变量名称,是要被处理的fasta文件,此处我们一行一行的迭代阅读这个巨大的fasta文件,line代表了每一行的字符串
for line in fasta:
#if 用于判断条件,.startswith(X)的意思是字符串以X开始
if line.startswith(">"):
#根据fasta的规定,>开始的行就是名称,我们去掉行里的"\n",这代表着换行
head = line.replace("\n","")
#向空字典dic这个容器中放入一个键值对,这可能比较抽象,可以想象成在固体颗粒上固定好了抗体准备吸附抗原了
dic[head] = ''
else:#esle表示了和它缩进相同的if的反面,这里就是字符串不以>开始,那么这一行应该是序列。
#去掉末尾的换行符
seq = line.replace("\n","")
#往键上赋值,+=代表了在之前的值后面续接。可以想象成这个抗原是个寡聚蛋白,抗体抓住抗原的一个亚基后,其他的亚基自己聚合上来。
dic[head] += seq
#循环结束以后输出dic,这个dic包含了一一对应的序列名与序列
return dic
#使用try进行异常控制
try:
#sys.argv[2]代表了python3后面第三个空格后输入的内容,此处应该是原始fasta文件
#open()用来打开文件,如果不存在就创建,“w+”表示覆盖原文件,创建一个空的,“a+”表示打开文件,光标移到最后一行
seqdump = open(sys.argv[2])
dic = fasta_2_dic(seqdump)
ofile = open(sys.argv[3],"w+")
ofile = open(sys.argv[3],"a+")
lst = open(sys.argv[1])
#迭代待分离的列表文件
for x in lst:
h = x.replace("\n","")
try:
#根据待分离文件中的名称(抗体),在字典中中找对应的序列(抗原)
seq = dic[h]
#对符合条件的序列输出打印到文件ofile
print(x+seq+"\n",file=ofile)
#如果没有该名称
except KeyError:
#显示该名称不存在于原始文件中
print(h+" not found in "+sys.argv[2])
#输出使用帮助
except IndexError:
print("Usage: python3 split.py <list> <fasta> <outfile>")
2.1.2 过滤过长或过短的序列
问题:我们在构建多序列比对的时候,为了比对的可靠性,需要过滤特别长或者特别短的序列。
方法:把fasta文件做成序列名称和序列对应的字典,求所有序列平均长度,输出长度符合范围的序列。
列表是Python中一种序列数据结构,是一种常见的数据类型,其中元素有自己从前到后,从0开始的索引(index),放在中括号内:
l = [element1,element2,element3]
numpy是一个广泛使用的用于一些计算的包,在求平均数等方面非常好用。
import sys
import numpy as np
#我们使用 def functionname(): 定义一个函数,方便调用。
def fasta_2_dic(fasta):
#创建一个名叫dic的空字典,我们将要建立序列名称和序列的一一对应关系。
dic = {}
#fasta是被传入函数的变量名称,是要被处理的fasta文件,此处我们一行一行的迭代阅读这个巨大的fasta文件,line代表了每一行的字符串
for line in fasta:
#if 用于判断条件,.startswith(X)的意思是字符串以X开始
if line.startswith(">"):
#根据fasta的规定,>开始的行就是名称,我们去掉行里的"\n",这代表着换行
head = line.replace("\n","")
#向空字典dic这个容器中放入一个键值对,这可能比较抽象,可以想象成在固体颗粒上固定好了抗体准备吸附抗原了
dic[head] = ''
else:#esle表示了和它缩进相同的if的反面,这里就是字符串不以>开始,那么这一行应该是序列。
#去掉末尾的换行符
seq = line.replace("\n","")
#往键上赋值,+=代表了在之前的值后面续接。可以想象成这个抗原是个寡聚蛋白,抗体抓住抗原的一个亚基后,其他的亚基自己聚合上来。
dic[head] += seq
#循环结束以后输出dic,这个dic包含了一一对应的序列名与序列
return dic
def _count(dic):
ofile = open(sys.argv[2],"w+")
ofile = open(sys.argv[2],"a+")
#空字典cdic
cdic = {}
#空列表clist
clist = []
#遍历字典dic中的每一个键key
for key in dic:
#len()返回字符串长度,列表元素数量,字典的键的个数,此处我们用dic[key]获得dic中储存的序列名对应的序列,并获得长度,将长度储存于变量seql中,seql应该代表一个整数,为长整型。
seql = len(dic[key])
#在字典cdic中储存序列名和序列长
cdic[key] = seql
#在clist中用append方法添加元素
clist.append(seql)
#此循环结束后,clist中储存了所有的序列长,cdic中储存了序列名和序列长的对应关系,dic中储存了序列名和序列的对应关系
#用numpy的mean()方法求clist所有元素的平均数
m = np.mean(clist)
#迭代dic
for key in dic:
#如果某序列的长度与所有序列平均值的比值大于0.8,小于1.2,则认为长度适中。
if 0.8 < cdic[key]/m < 1.2:
#对符合条件的序列输出打印到文件ofile
print(key+"\n"+dic[key]+"\n",file=ofile)
try:
seqdump = open(sys.argv[1])
dic = fasta_2_dic(seqdump)
_count(dic)
except IndexError:
print("Usage: python3 fliter.py <sequences.fasta> <out file>")
功能基因组学
BLAST,序列搜索第一步
生物信息学中,BLAST(英语:Basic Local Alignment Search Tool)是一个用来比对生物序列的一级结构(如不同蛋白质的氨基酸序列或不同基因的DNA序列)的算法。 已知一个包含若干序列的数据库,BLAST可以让你在其中寻找与其感兴趣的序列相同或类似的序列。
BLAST在线版,生物序列的Google
问题:201X年,A组报道了B蛋白有降解PET(一种塑料)的活性,发了science。你很感兴趣,希望找到一个不一样的C蛋白也能水解PET,可以发AMB保毕业。
流程:获取B蛋白序列,BLAST。
- 寻找B蛋白序列,获得的序列如下:
此处的序列为fasta格式,一般来说,fasta格式中以>开头的行是序列的名称物种等信息,接下来直到下一个>之间的均为序列,60或80个字母为一行,但序列部分的分行不重要。
> sp|A0A0K8P6T7|PETH_IDESA Poly(ethylene terephthalate) hydrolase MNFPRASRLMQAAVLGGLMAVSAAATAQTNPYARGPNPTAASLEASAGPFTVRSFTVSRPSGYGAGTVY YPTNAGGTVGAIAIVPGYTARQSSIKWWGPRLASHGFVVITIDTNSTLDQPSSRSSQQMAALRQVASLN GTSSSPIYGKVDTARMGVMGWSMGGGGSLISAANNPSLKAAAPQAPWDSSTNFSSVTVPTLIFACENDS IAPVNSSALPIYDSMSRNAKQFLEINGGSHSCANSGNSNQALIGKKGVAWMKRFMDNDTRYSTFACENP NSTRVSDFRTANCS
- BLAST search:
访问NCBI BLAST或UniProt BLAST,一般我在搜索核酸序列时会使用NCBI,搜索蛋白序列时使用UniProt,这两个数据库其实是互联互通的。以下介绍UniProt的使用。
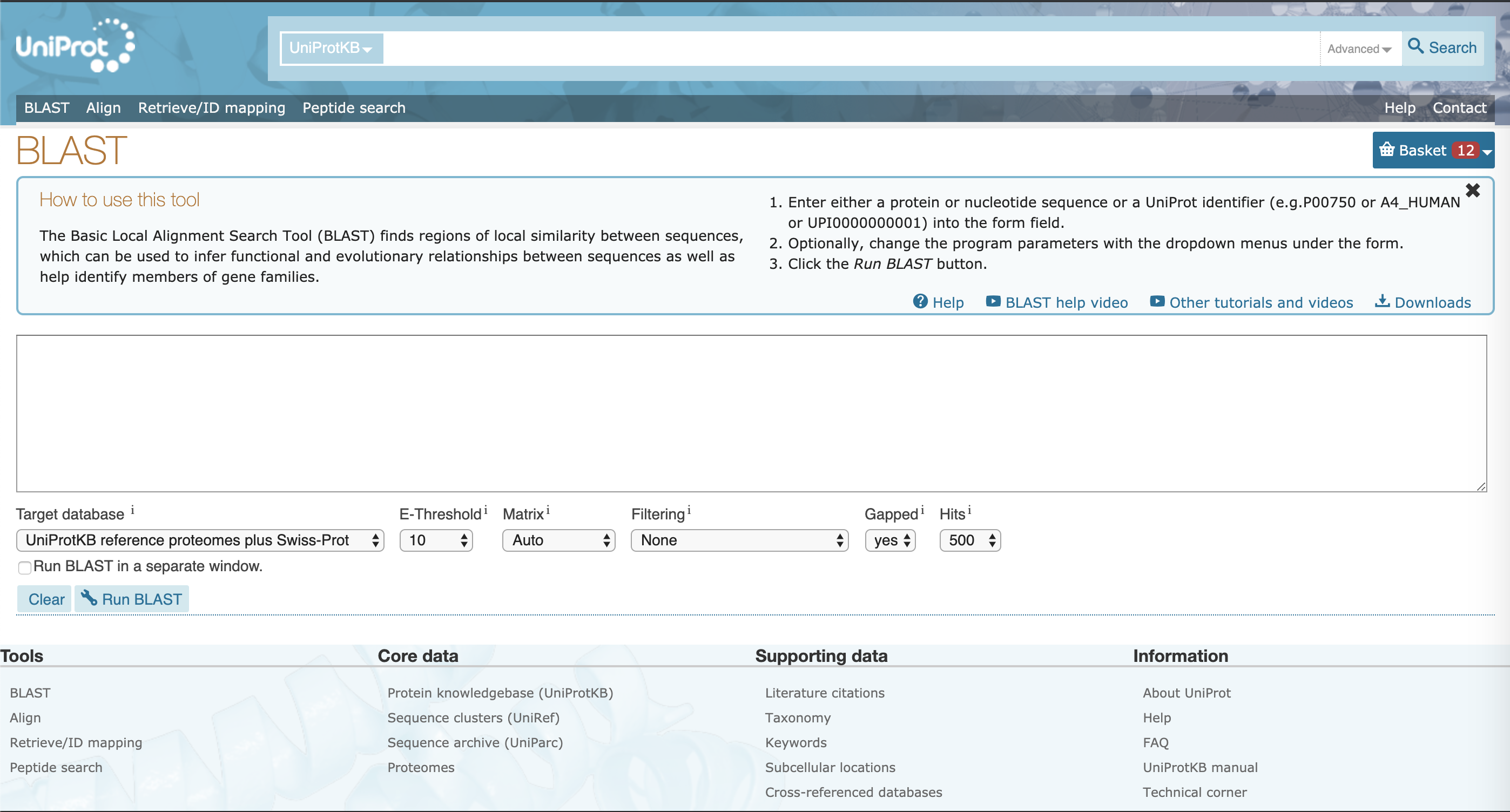 最上面是搜索框,对于这条蛋白序列,只要在搜索框内输入
最上面是搜索框,对于这条蛋白序列,只要在搜索框内输入A0A0K8P6T7进行搜索即可获得详细的信息。
将fasta格式的序列或者序列号A0A0K8P6T7 输入进大文本框,点击Run BLAST,即可运行BLAST。
| 名称 | 含义 |
|---|---|
| Target database | 被搜索的数据库,UniProtKB referance proteomes plus Swiss-Prot是最大最全的数据库,常用的还有Swiss-Prot,是所有人工注释的序列;PDB是所有的晶体结构的序列。 |
| E-Threshold | E值的上限,两个蛋白质序列E值越小,同源的可能性愈大。 |
| Matrix | 打分矩阵,一般使用Auto,在输入序列存在较少同源序列,要寻找亲缘关系较远的序列时可使用BLOSUM-45,在输入序列存在较多同源序列,要寻找亲缘关系较近的序列时使用BLOSUM-80。 |
| Hits | 返回结果的个数 |
输出的结果如下:
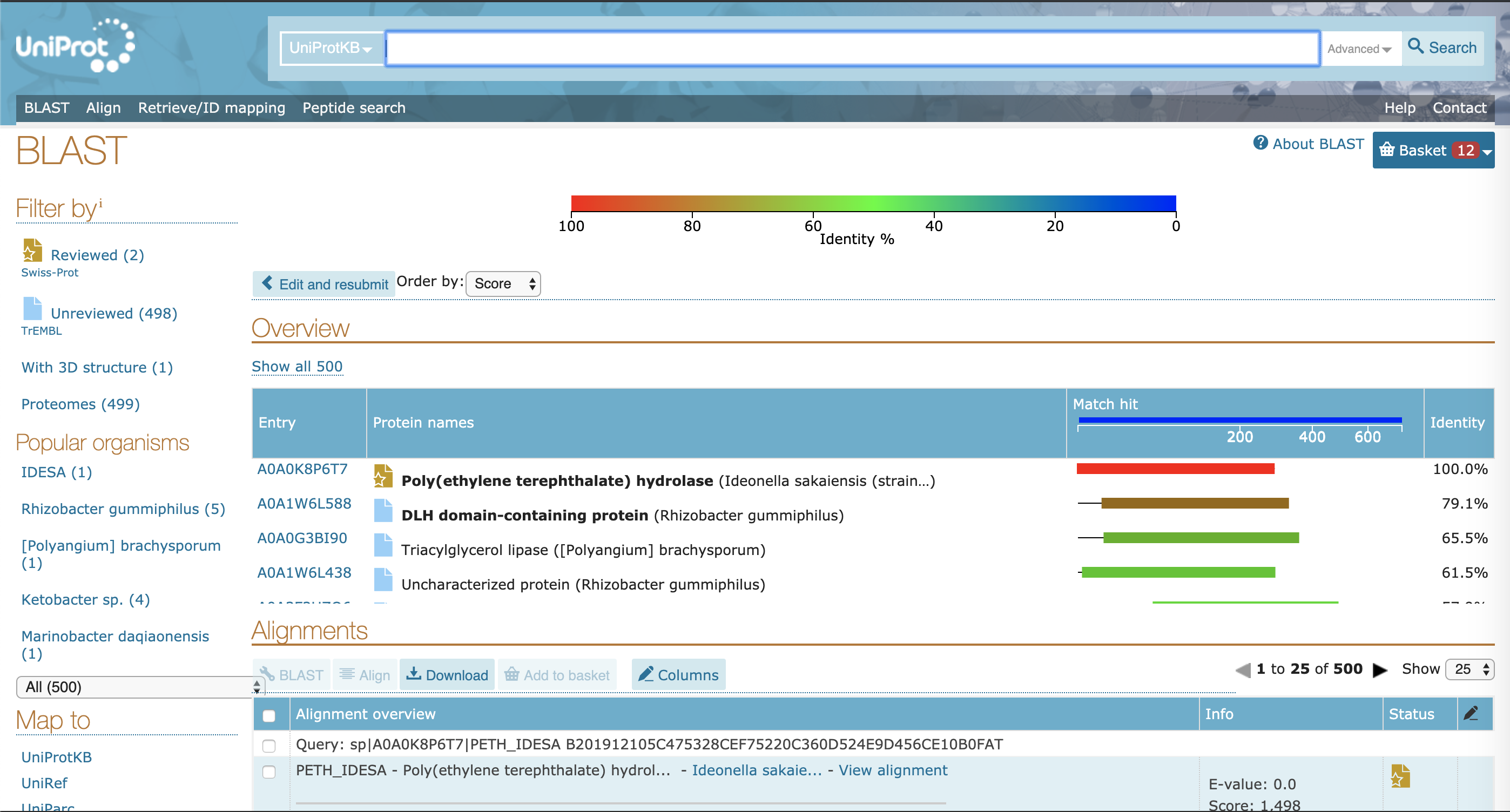
在Alignment部分,可以在感兴趣的条目中点击View alignment,查看两条序列的比对情况,一般来说Identity大于50%的序列就有可能拥有相似的功能,对于发育关系较远的蛋白,比如植物合成某些代谢产物的蛋白在细菌中的同源蛋白可能相似度只有30%。这里我们观察一下第二个结果A0A1W6L588 ,来自 Rhizobacter gummiphilus 的 DLH domain-containing protein,简称DLH。序列比对情况如下:
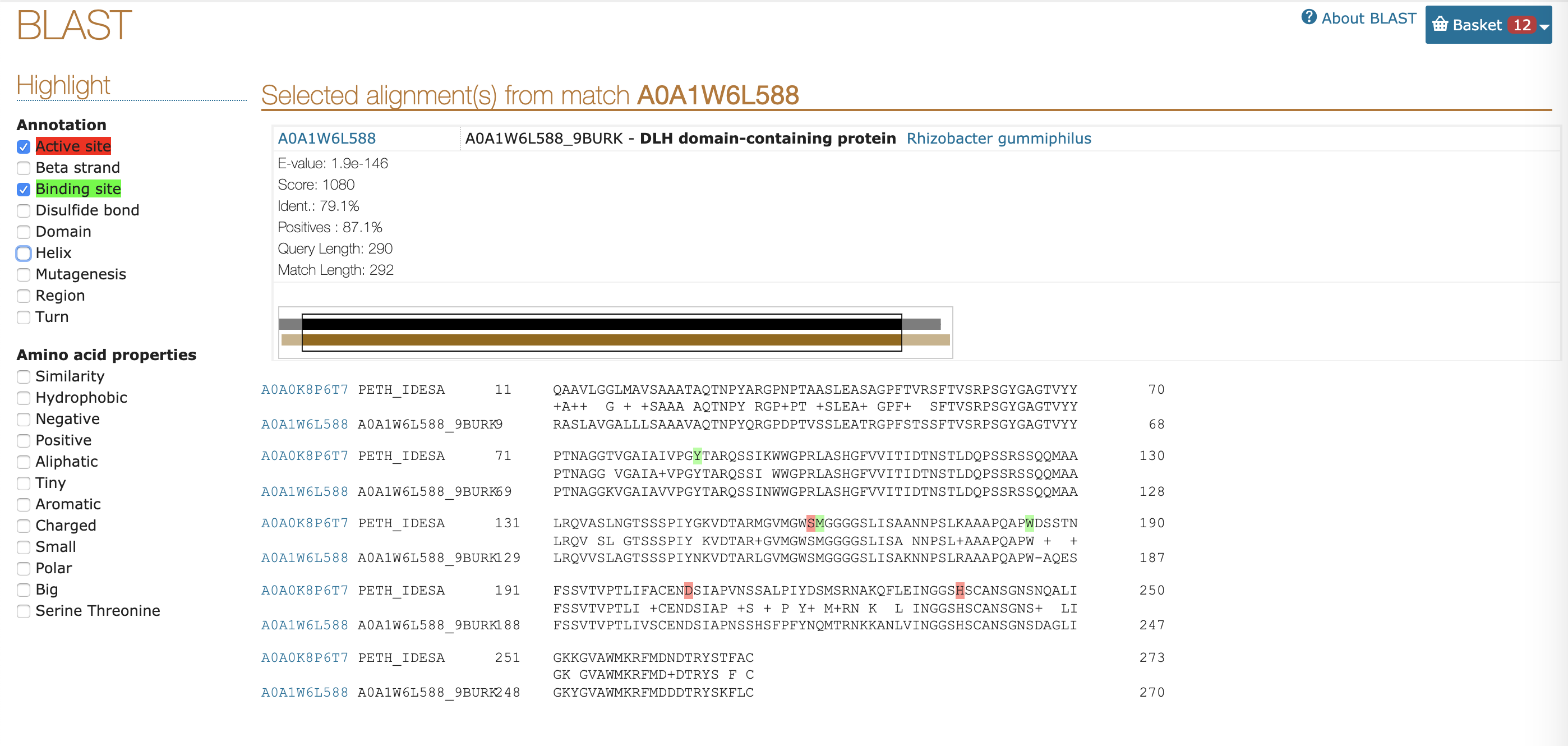 从序列比对中,我们可以发现,DLH与PETase的底物结合位点和催化三联体均保守,推测DLH同样可以水解PET。
从序列比对中,我们可以发现,DLH与PETase的底物结合位点和催化三联体均保守,推测DLH同样可以水解PET。
BLAST的本地版,你和菌菌的秘密花园
问题:201X年,A组报道B菌有降解有机物C的活性,在数据库中可以查到C的降解方式,且B菌基因组已经被测序。
流程:获取B菌基因组,确定降解通路中涉及蛋白质的序列,安装本地版BLAST软件,BLAST。
BLAST本地版安装
访问NCBI的BLAST网站,下载可执行程序(点击访问ftp站点)
NCBI的速度一向不行,所以我们要在服务器上先安装高速下载工具axel:
sudo apt-get install axel
axel 中的 - n 可以设置连接数,不建议过大,会导致NCBI封禁你的IP地址。 此处下载的是linux版本的BLAST 2.9.0, 支持V5数据库。
axel -n 20 ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.9.0+-x64-linux.tar.gz
解压文件:
tar -vxzf ncbi-blast-2.9.0+-x64-linux.tar.gz
将文件拷贝进/opt:
sudo cp -r ncbi-blast-2.9.0+/opt
添加路径至.bashrc文件:
vi ~/.bashrc
确保键盘处于英文输入状态,按下shift + G,定位到文件最后一行,然后按下o,添加:
export PATH="$PATH:/opt/ncbi-blast-2.9.0+/bin:"
按下ESC,按住shift连敲两下z键,保存退出文件。
source ~/.bashrc
此时,输入blastp -version,回车,应当可以看见:
blastp: 2.9.0+
Package: blast 2.9.0, build Mar 11 2019 15:20:05
代表安装成功。
- 有报道称 Phialophora americana 可以降解
Zearalenone (ZEN),已知ZEN lactonase (ZHD101)的序列,在NCBI中可以查询到 P. americana 的完整蛋白质组。 - 分别下载
ZHD101和 P. americana 全部蛋白质序列,分别保存为Q8NKB0.fasta和Pa.fasta。
wget https://www.uniprot.org/uniprot/Q8NKB0.fasta
axel -n 10 ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/835/435/GCA_000835435.1_Capr_semi_CBS27337_V1/GCA_000835435.1_Capr_semi_CBS27337_V1_protein.faa.gz
gunzip GCA_000835435.1_Capr_semi_CBS27337_V1_protein.faa.gz
mv GCA_000835435.1_Capr_semi_CBS27337_V1_protein.faa Pa.fasta
- 建立本地 P. americana 数据库:
makeblastdb -in Pa.fasta -dbtype prot -out Pa
- 运行本地BLAST:
blastp -query Q8NKB0.fasta -db Pa > zhd101_Pa.txt
本例的现实意义在于:你的课题组筛到了菌,测了基因组,要在里面找这个蛋白序列,你不能提前上传到NCBI,只能自己本地BLAST。
多序列比对与进化树,今天我们都是达尔文
问题:对于一组序列,需要对其并观察保守位点,产生MSA (multiple sequence alignment) 文件,绘制进化树。
流程:准备fasta文件,安装muscle,产生比对文件,安装FastTree,构建进化树,iTOL在线可视化。
需求:Ubuntu
- 下载一组
globin的蛋白序列:
wget https://raw.githubusercontent.com/EddyRivasLab/hmmer/master/tutorial/globins45.fa
- 安装
muscle:
sudo apt-get install muscle
- 运行
muscle产生适合观察的CLUSTALW格式:
muscle -in globins45.fa -out globins45.clw -clw
产生适合其他软件读取的fasta格式:
muscle -in globins45.fa -out globins45.ali
- 下载安装
FastTree,前面安装BLAST时,已经将 /opt/ncbi-blast-2.9.0+/bin 添入~/.bashrc:
wget http://www.microbesonline.org/fasttree/FastTree
chmod +x FastTree
sudo cp FastTree /opt/ncbi-blast-2.9.0+/bin/
- 构建进化树:
FastTree globins45.ali > globins45.tree
- 将
globins45.tree文件上传至iTOL在线可视化
HMMER,比BLAST更进一步
背景:之前,讨论了如何利用BLAST搜索同源蛋白。对于一个蛋白质家族,其中如果有多条实验验证过功能的序列,同时序列之间差异较大,无法选择合适的一条序列时,使用HMMER进行搜索。HMMER已经展现了比BLAST更高的准确率,正在变得更加普及。
问题:糖苷水解酶是一类广泛分布的水解酶,糖苷水解酶家族非常庞大,序列复杂性非常高,纤维素酶(3.2.1.4)通常属于GH5,但是在GH11、GH45等家族中也有分布,同样的,GH5家族内也有许多酶不属于3.2.1.4。我们搜索C真菌内是否有GH5家族的纤维素酶。
流程:获得C的所有蛋白质序列,构建GH5纤维素酶的隐普马尔科夫模型,使用HMMER搜索。
- 下载C蛋白序列(和本地BLAST教程使用的是同一物种)
axel -n 10 ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/835/435/GCA_000835435.1_Capr_semi_CBS27337_V1/GCA_000835435.1_Capr_semi_CBS27337_V1_protein.faa.gz
gunzip GCA_000835435.1_Capr_semi_CBS27337_V1_protein.faa.gz
mv GCA_000835435.1_Capr_semi_CBS27337_V1_protein.faa Pa.fasta
- 安装HMMER:
sudo apt-get install hmmer
- 下载GH5纤维素酶序列,这些序列是我从Swiss-Prot里下载的总共30条仅包含GH5的纤维素酶:
wget 139.224.116.102/GH5_3_2_1_4.fasta
- 多序列比对:
HMM profile的准确率严重依赖于多序列比对的正确性,而多序列比对有意义的前提在于被对齐的序列首先在高级结构上是一致的。所以我写了一个小脚本,限制序列长度的范围在平均值的(0.8, 1.2)之内。
wget 139.224.116.102/filter.py
python3 filter.py GH5_3_2_1_4.fasta GH5_3_2_1_4.fas
muscle -in GH5_3_2_1_4.fas -out GH5_3_2_1_4.ali
- 构建HMM profile:
hmmbuild GH5_3_2_1_4.hmm GH5_3_2_1_4.ali
- 用HMM profile给序列打分:
hmmsearch GH5_3_2_1_4.hmm Pa.fasta
对于P. americana 似乎没有明显的属于GH5的cellulase。P10477没有包含在过滤之后建HMM的序列里,因为长度太长了,GH5_3_2_1_4.hmm对P10477打分为327.8。下图为hmmsearch结果:
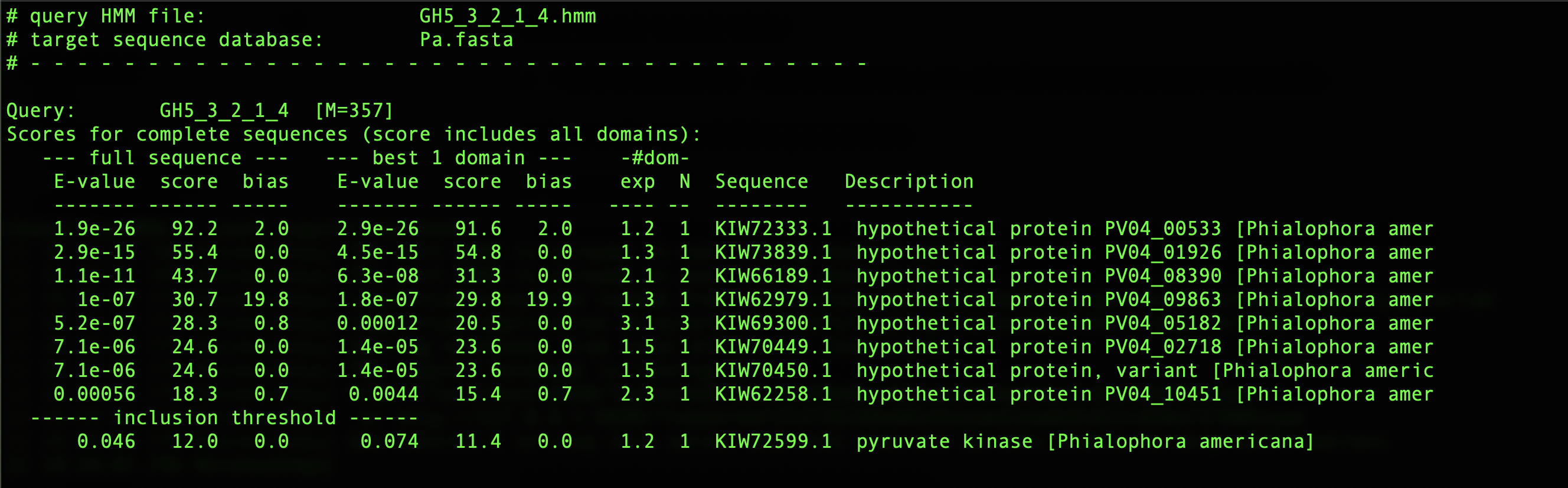
HMM简介
InterproScan,更大更全
InterPro通过将蛋白质分类为家族并预测域和重要位点来提供蛋白质的功能分析。 InterPro结合所有权威的数据库包括pfam,cdd,gene3d,prosite等15个数据库,非常适合预测蛋白质的结构域。
对一条蛋白质序列进行手动注释
背景:在HMMER的例子中,我们发现了
KIW72333.1这条机器注释为hypothetical protein的序列,下面我们介绍一下怎么样使用这几个数据库和软件完成注释。问题:对于一条测序得到的蛋白序列,使用生信手段初步预测其大致功能。
流程:结合BLAST、HMMER和InterPro,推测其生物学功能。
- 获取序列
KIW72333.1:
>KIW72333.1 hypothetical protein PV04_00533 [Phialophora americana]
MATGILKVKGTQVVGNDGKPVILRGCAIGGWLNMENFIVGYPGHESSIRAAMLAAMGQENYDFFFDRWLYYFFTEADAKF
FKSMGLNCIRIPFNYRHFEDDMNPRVLKESGFKHLDRVIDLCAKEGIYTILDMHTVPGGQGPGWHADNTTSYAAFWDYKD
HQDRTVWLWEQLAQRYKGNPWIAGYNPINEPCDPKHVRLPAFYARFEKAIRAIDPDHILWLDGNTFAAEWKGFDTVLPNC
VYALHDYSMMGFPTGRPYEGTAEQKERLERQFLRKSEFQRTHNTAIWNGEFGPVYANPKWDENAEEVNQKRYNMLGEQLR
IYDKFQIPWSIWLWKDVGLQGMVYTSPDSAWNKLIEPILEKKKRLQLDAWGKYPSKEVEDLIQPLVSWIDSVSPTAKDVY
PSTWNTARHIERQVLQTFLAETFCQEFAELFRDKDQAALEELAKSFSFENCVQRDGLNKIMSDYAAIATSSGGK
- BLAST搜索Swiss-Prot数据库
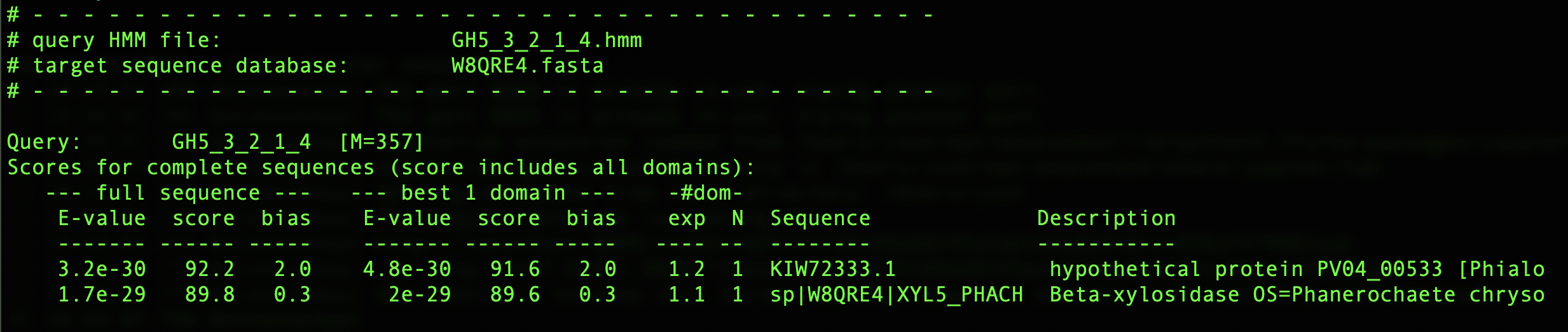
发现其与一个木糖酶(3.2.1.37)W8QRE4有极高的同源性,且此木聚糖酶属于GH5家族且没有纤维素酶(3.2.1.4)活性。
- InterPro搜索
发现其被注释为GH5家族,GH5家族常见纤维素酶(3.2.1.4)
- 本地HMM搜索
发现 KIW72333.1与GH5的纤维素酶相似度不高,和W8QRE4与GH5的纤维素酶相似度相仿。
- 推测,
KIW72333.1有可能是一个少见的属于GH5家族的木聚糖酶,有可能广泛存在着属于GH5的木聚糖酶,下面的思路是可以分析一下整个GH5家族其他的蛋白序列,看看有没有不明显属于纤维素酶但和木聚糖酶比较相似的。
结构生物信息学
结构生物信息学是建立在结构生物学和生物信息学(广义的生物信息学,包括了informatics和computation)之上的一门学科,这里我觉得有必要回顾一下历史。
1840年,英国人的坚船利炮轰开了清王朝的大门,同时,德国化学家第一次将血红蛋白描述为 “血晶” 。
1895年,李中堂乘坐德国商船“公义号”远赴日本,与此同时,德国人威廉·康拉德·伦琴 (Wilhelm Conrad Röntgen) 发现了X射线 (X-rays)。
1912年,中华民国正式结束了清王朝,同年劳厄使用X射线照射硫酸铜晶体,获得了规律性的图样。1915年布拉格父子和劳厄共同获得诺贝尔物理学奖,布拉格定律和X射线衍射在此后的一个多世纪里被广泛使用。
1958年,中华人民共和国迎来三年自然灾害,英国科学家约翰·肯德鲁用剑桥的EDSAC Mark I电子计算机上解析了抹香鲸肌红蛋白的三维结构。
1993年,邓小平已经完成了南巡讲话,人大将“社会主义初级阶段”写入宪法。Andrej Šali发表《Comparative Protein Modelling by Satisfaction of Spatial Restraints》,modeller问世。Google Scholar显示,该文章目前已被引用11659次,中国的GDP也已经是当年世界第二日本的两倍。
2020年,世纪被疫情笼罩,迎来百年未有之大变局,AlphaFold也掀起了结构生物信息学的革命,结构预测达到了和实验一致的精度。
同源建模
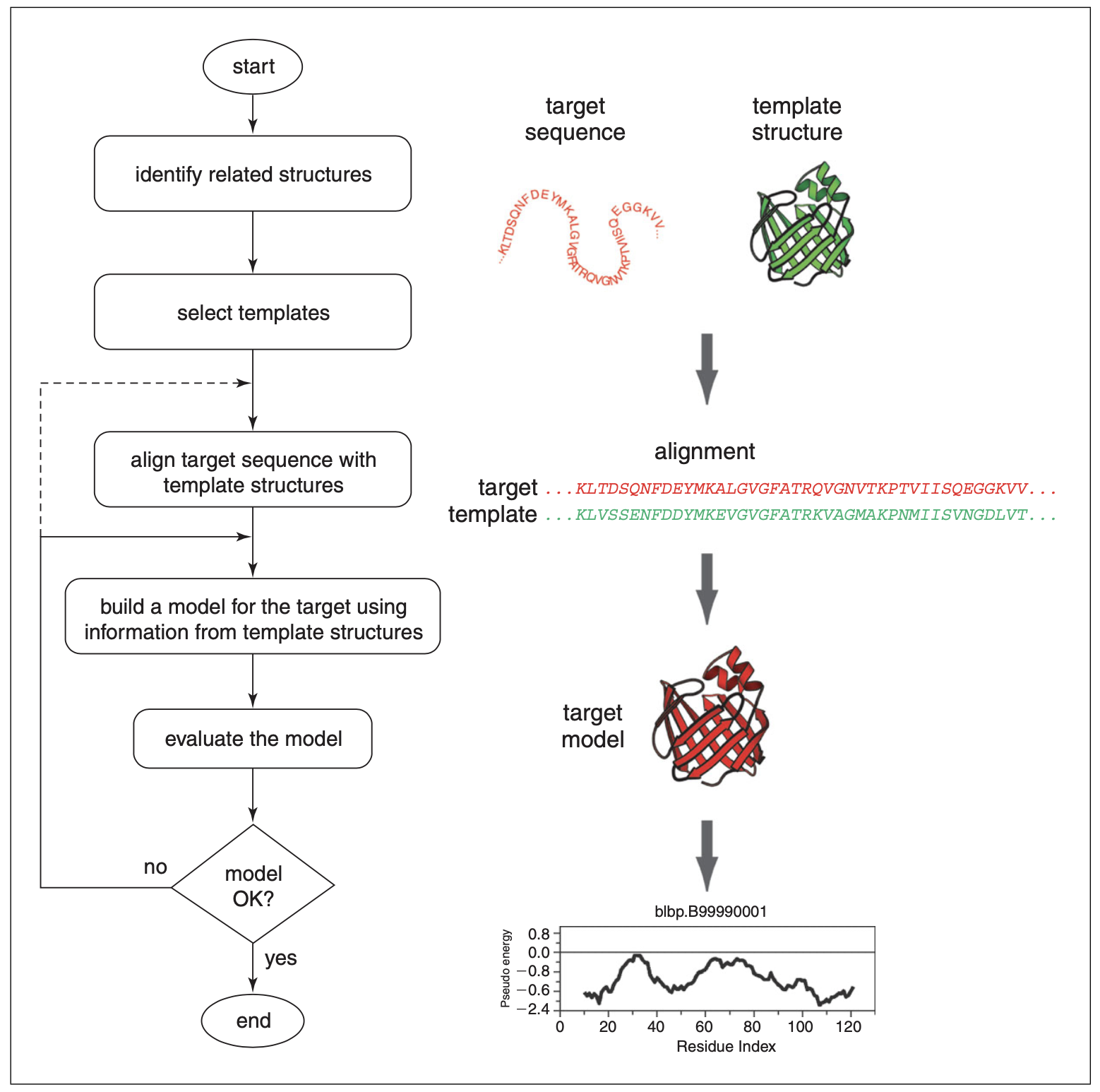
在生物学中,蛋白质序列功能的表征是最为常见的问题,而这一项任务通常需要该蛋白准确的三维结构。在没有实验获得的三维结构时,同源建模可以根据至少一个已知结构的相似的蛋白提供一个可用的三维模型。同源建模通常包括这四步:1. 折叠识别 (fold assignment) ,为目标蛋白 (target) 寻找同源的模版蛋白 (template) ,这一步通常使用BLAST完成。2. 将目标和模版对齐 (alignment) 。3. 根据对齐的模版创建模型。4. 验证。
-
下载并安装Modeller,Modeller是一个学术开源软件,首先到注册页面注册,邮箱会收到license key。然后去下载页面下载自己系统所需的安装文件进行安装,本文的教程是以Ubuntu系统为基础开展的,其他系统的安装方法在modeller官网的installation里也有详细描述。
下载deb安装文件并安装:
wget https://salilab.org/modeller/9.23/modeller_9.23-1_amd64.deb
sudo env KEY_MODELLER=<此处是你的license key> dpkg -i modeller_9.23-1_amd64.deb
- 获取2019-nCoV的S protein序列,以下简称2019S,并准备成modeller输入序列的固定格式,保存为2019S.ali:
>P1;2019S sequence:2019S:::::::0.00: 0.00 MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV SGTNGTKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGTTLDSKTQSLLIVNNATNVVIKVCEFQFCNDPF LGVYYHKNNKSWMESEFRVYSSANNCTFEYVSQPFLMDLEGKQGNFKNLREFVFKNIDGYFKIYSKHTPI NLVRDLPQGFSALEPLVDLPIGINITRFQTLLALHRSYLTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYN ENGTITDAVDCALDPLSETKCTLKSFTVEKGIYQTSNFRVQPTESIVRFPNITNLCPFGEVFNATRFASV YAWNRKRISNCVADYSVLYNSASFSTFKCYGVSPTKLNDLCFTNVYADSFVIRGDEVRQIAPGQTGKIAD YNYKLPDDFTGCVIAWNSNNLDSKVGGNYNYLYRLFRKSNLKPFERDISTEIYQAGSTPCNGVEGFNCYF PLQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVCGPKKSTNLVKNKCVNFNFNGLTGTGVLTESNKKFL PFQQFGRDIADTTDAVRDPQTLEILDITPCSFGGVSVITPGTNTSNQVAVLYQDVNCTEVPVAIHADQLT PTWRVYSTGSNVFQTRAGCLIGAEHVNNSYECDIPIGAGICASYQTQTNSPRRARSVASQSIIAYTMSLG AENSVAYSNNSIAIPTNFTISVTTEILPVSMTKTSVDCTMYICGDSTECSNLLLQYGSFCTQLNRALTGI AVEQDKNTQEVFAQVKQIYKTPPIKDFGGFNFSQILPDPSKPSKRSFIEDLLFNKVTLADAGFIKQYGDC LGDIAARDLICAQKFNGLTVLPPLLTDEMIAQYTSALLAGTITSGWTFGAGAALQIPFAMQMAYRFNGIG VTQNVLYENQKLIANQFNSAIGKIQDSLSSTASALGKLQDVVNQNAQALNTLVKQLSSNFGAISSVLNDI LSRLDKVEAEVQIDRLITGRLQSLQTYVTQQLIRAAEIRASANLAATKMSECVLGQSKRVDFCGKGYHLM SFPQSAPHGVVFLHVTYVPAQEKNFTTAPAICHDGKAHFPREGVFVSNGTHWFVTQRNFYEPQIITTDNT FVSGNCDVVIGIVNNTVYDPLQPELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVA KNLNESLIDLQELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD SEPVLKGVKLHYT*
需要注意的是,第一行>P1;之后的部分为这个蛋白的名字,第二行是sequence开头,第一和第二个冒号中间是你的蛋白的名字。其余部分不需要改动。第三行开始是序列,和fasta类似但是末尾要加一个*。
- 利用BLAST工具查找PDB数据库S的同源蛋白,参考BLSAT,确定模版结构

从结果中我们可以发现,2019S与来自SRAS的突刺蛋白有非常高的同源性,identity在75%左右。我们需要在这些不同的晶体结构中找一个最适合的作为同源建模的模版。但是本教程会把这些都作为模版建模,我们可以从产生的结果中看出模版选择对模型质量的重要意义。

- 将2019S与模版对齐,与单纯的序列比对不同,这里的对齐参考了结构的信息,使用可变间隙补偿函数 (variable gap penalty function) 将间隙放置在溶剂暴露且弯曲、二级结构之外以及空间上靠近的两个位置之间。与标准序列比对技术相比,比对误差减少了约三分之一。随着序列之间相似性的降低和空位数量的增加,这种改进变得更加重要。
#此为文件align2d.py
from modeller import *
env = environ()
aln = alignment(env)
mdl = model(env, file='6CRV', model_segment=('FIRST:A','LAST:A'))#6CRV是模版的名字,A表示A链
aln.append_model(mdl, align_codes='6CRVA', atom_files='6CRV.pdb')#atom_file是pdb文件的名字
aln.append(file='2019S.ali', align_codes='2019S')
aln.align2d()
aln.write(file='2019S-6CRVA.ali', alignment_format='PIR')
aln.write(file='2019S-6CRVA.pap', alignment_format='PAP')
运行比对,由于序列太长,这个比对耗时比较长:
mod9.23 align2d.py
from modeller import *
from modeller.automodel import *
#from modeller import soap_protein_od
env = environ()
a = automodel(env, alnfile='2019S-6CRVA.ali',
knowns='6CRVA', sequence='2019S',
assess_methods=(assess.DOPE,
#soap_protein_od.Scorer(),
assess.GA341))
a.starting_model = 1
a.ending_model = 5
a.make()
from modeller import *
from modeller.scripts import complete_pdb
log.verbose() # request verbose output
env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology
env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# read model file
mdl = complete_pdb(env, '2019S.B99990002.pdb')
# Assess with DOPE:
s = selection(mdl) # all atom selection
s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='2019S.profile',
normalize_profile=True, smoothing_window=15)
import pylab
import modeller
def get_profile(profile_file, seq):
"""Read `profile_file` into a Python array, and add gaps corresponding to
the alignment sequence `seq`."""
# Read all non-comment and non-blank lines from the file:
f = file(profile_file)
vals = []
for line in f:
if not line.startswith('#') and len(line) > 10:
spl = line.split()
vals.append(float(spl[-1]))
# Insert gaps into the profile corresponding to those in seq:
for n, res in enumerate(seq.residues):
for gap in range(res.get_leading_gaps()):
vals.insert(n, None)
# Add a gap at position '0', so that we effectively count from 1:
vals.insert(0, None)
return vals
e = modeller.environ()
a = modeller.alignment(e, file='2019S-6CRVA.ali')
template = get_profile('6CRVA.profile', a['6CRVA'])
model = get_profile('2019S.profile', a['2019S'])
# Plot the template and model profiles in the same plot for comparison:
pylab.figure(1, figsize=(10,6))
pylab.xlabel('Alignment position')
pylab.ylabel('DOPE per-residue score')
pylab.plot(model, color='red', linewidth=2, label='Model')
pylab.plot(template, color='green', linewidth=2, label='Template')
pylab.legend()
pylab.savefig('dope_profile.png', dpi=65)