结构生物信息学
结构生物信息学是建立在结构生物学和生物信息学(广义的生物信息学,包括了informatics和computation)之上的一门学科,这里我觉得有必要回顾一下历史。
1840年,英国人的坚船利炮轰开了清王朝的大门,同时,德国化学家第一次将血红蛋白描述为 “血晶” 。
1895年,李中堂乘坐德国商船“公义号”远赴日本,与此同时,德国人威廉·康拉德·伦琴 (Wilhelm Conrad Röntgen) 发现了X射线 (X-rays)。
1912年,中华民国正式结束了清王朝,同年劳厄使用X射线照射硫酸铜晶体,获得了规律性的图样。1915年布拉格父子和劳厄共同获得诺贝尔物理学奖,布拉格定律和X射线衍射在此后的一个多世纪里被广泛使用。
1958年,中华人民共和国迎来三年自然灾害,英国科学家约翰·肯德鲁用剑桥的EDSAC Mark I电子计算机上解析了抹香鲸肌红蛋白的三维结构。
1993年,邓小平已经完成了南巡讲话,人大将“社会主义初级阶段”写入宪法。Andrej Šali发表《Comparative Protein Modelling by Satisfaction of Spatial Restraints》,modeller问世。Google Scholar显示,该文章目前已被引用11659次,中国的GDP也已经是当年世界第二日本的两倍。
2020年,世纪被疫情笼罩,迎来百年未有之大变局,AlphaFold也掀起了结构生物信息学的革命,结构预测达到了和实验一致的精度。
同源建模
在生物学中,蛋白质序列功能的表征是最为常见的问题,而这一项任务通常需要该蛋白准确的三维结构。在没有实验获得的三维结构时,同源建模可以根据至少一个已知结构的相似的蛋白提供一个可用的三维模型。同源建模通常包括这四步:1. 折叠识别 (fold assignment) ,为目标蛋白 (target) 寻找同源的模版蛋白 (template) ,这一步通常使用BLAST完成。2. 将目标和模版对齐 (alignment) 。3. 根据对齐的模版创建模型。4. 验证。
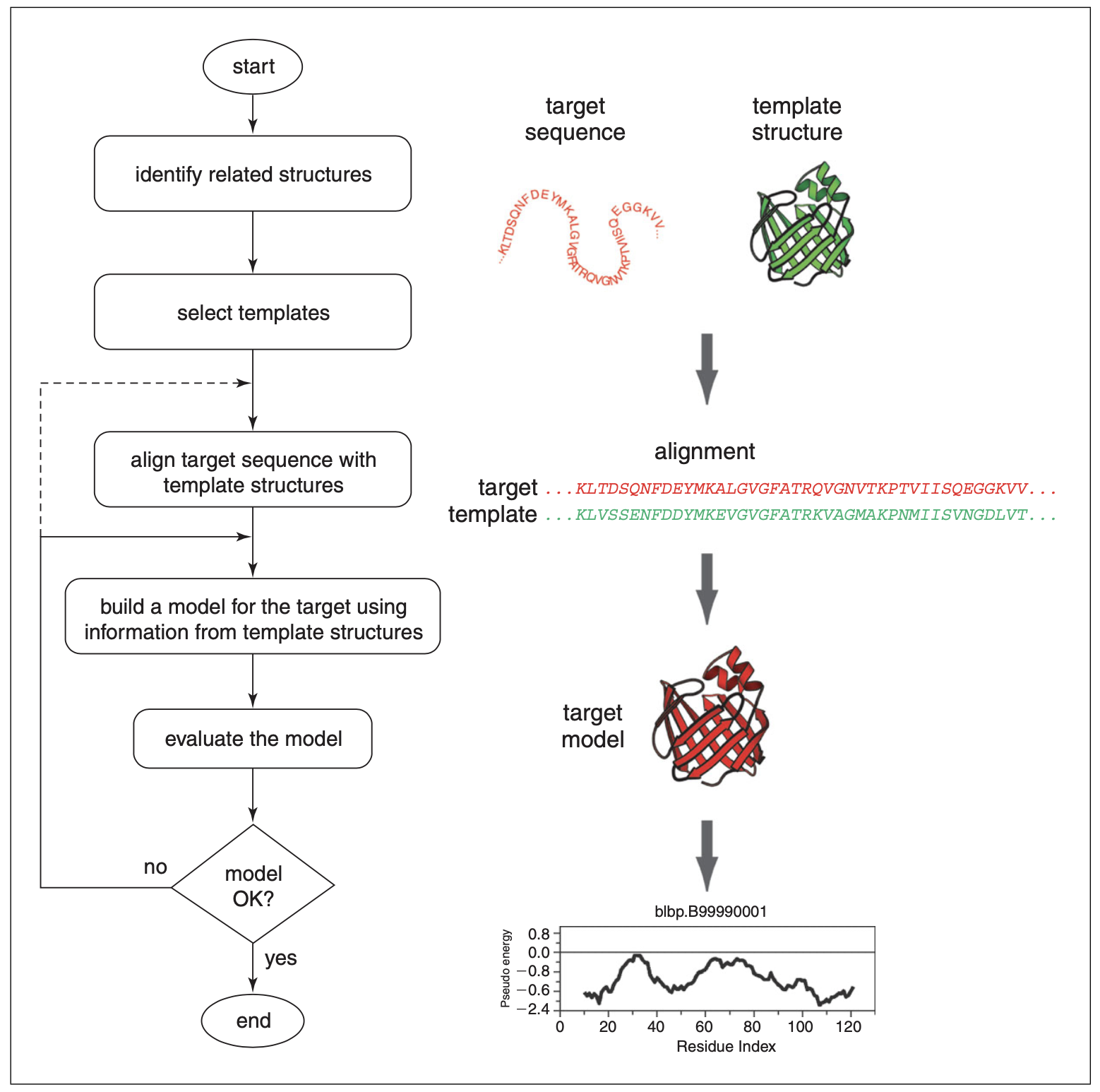
-
下载并安装Modeller,Modeller是一个学术开源软件,首先到注册页面注册,邮箱会收到license key。然后去下载页面下载自己系统所需的安装文件进行安装,本文的教程是以Ubuntu系统为基础开展的,其他系统的安装方法在modeller官网的installation里也有详细描述。
下载deb安装文件并安装:
wget https://salilab.org/modeller/9.23/modeller_9.23-1_amd64.deb
sudo env KEY_MODELLER=<此处是你的license key> dpkg -i modeller_9.23-1_amd64.deb
- 获取2019-nCoV的S protein序列,以下简称2019S,并准备成modeller输入序列的固定格式,保存为2019S.ali:
>P1;2019S sequence:2019S:::::::0.00: 0.00 MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV SGTNGTKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGTTLDSKTQSLLIVNNATNVVIKVCEFQFCNDPF LGVYYHKNNKSWMESEFRVYSSANNCTFEYVSQPFLMDLEGKQGNFKNLREFVFKNIDGYFKIYSKHTPI NLVRDLPQGFSALEPLVDLPIGINITRFQTLLALHRSYLTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYN ENGTITDAVDCALDPLSETKCTLKSFTVEKGIYQTSNFRVQPTESIVRFPNITNLCPFGEVFNATRFASV YAWNRKRISNCVADYSVLYNSASFSTFKCYGVSPTKLNDLCFTNVYADSFVIRGDEVRQIAPGQTGKIAD YNYKLPDDFTGCVIAWNSNNLDSKVGGNYNYLYRLFRKSNLKPFERDISTEIYQAGSTPCNGVEGFNCYF PLQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVCGPKKSTNLVKNKCVNFNFNGLTGTGVLTESNKKFL PFQQFGRDIADTTDAVRDPQTLEILDITPCSFGGVSVITPGTNTSNQVAVLYQDVNCTEVPVAIHADQLT PTWRVYSTGSNVFQTRAGCLIGAEHVNNSYECDIPIGAGICASYQTQTNSPRRARSVASQSIIAYTMSLG AENSVAYSNNSIAIPTNFTISVTTEILPVSMTKTSVDCTMYICGDSTECSNLLLQYGSFCTQLNRALTGI AVEQDKNTQEVFAQVKQIYKTPPIKDFGGFNFSQILPDPSKPSKRSFIEDLLFNKVTLADAGFIKQYGDC LGDIAARDLICAQKFNGLTVLPPLLTDEMIAQYTSALLAGTITSGWTFGAGAALQIPFAMQMAYRFNGIG VTQNVLYENQKLIANQFNSAIGKIQDSLSSTASALGKLQDVVNQNAQALNTLVKQLSSNFGAISSVLNDI LSRLDKVEAEVQIDRLITGRLQSLQTYVTQQLIRAAEIRASANLAATKMSECVLGQSKRVDFCGKGYHLM SFPQSAPHGVVFLHVTYVPAQEKNFTTAPAICHDGKAHFPREGVFVSNGTHWFVTQRNFYEPQIITTDNT FVSGNCDVVIGIVNNTVYDPLQPELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVA KNLNESLIDLQELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD SEPVLKGVKLHYT*
需要注意的是,第一行>P1;之后的部分为这个蛋白的名字,第二行是sequence开头,第一和第二个冒号中间是你的蛋白的名字。其余部分不需要改动。第三行开始是序列,和fasta类似但是末尾要加一个*。
- 利用BLAST工具查找PDB数据库S的同源蛋白,参考BLSAT,确定模版结构

从结果中我们可以发现,2019S与来自SRAS的突刺蛋白有非常高的同源性,identity在75%左右。我们需要在这些不同的晶体结构中找一个最适合的作为同源建模的模版。但是本教程会把这些都作为模版建模,我们可以从产生的结果中看出模版选择对模型质量的重要意义。

- 将2019S与模版对齐,与单纯的序列比对不同,这里的对齐参考了结构的信息,使用可变间隙补偿函数 (variable gap penalty function) 将间隙放置在溶剂暴露且弯曲、二级结构之外以及空间上靠近的两个位置之间。与标准序列比对技术相比,比对误差减少了约三分之一。随着序列之间相似性的降低和空位数量的增加,这种改进变得更加重要。
#此为文件align2d.py
from modeller import *
env = environ()
aln = alignment(env)
mdl = model(env, file='6CRV', model_segment=('FIRST:A','LAST:A'))#6CRV是模版的名字,A表示A链
aln.append_model(mdl, align_codes='6CRVA', atom_files='6CRV.pdb')#atom_file是pdb文件的名字
aln.append(file='2019S.ali', align_codes='2019S')
aln.align2d()
aln.write(file='2019S-6CRVA.ali', alignment_format='PIR')
aln.write(file='2019S-6CRVA.pap', alignment_format='PAP')
运行比对,由于序列太长,这个比对耗时比较长:
mod9.23 align2d.py
from modeller import *
from modeller.automodel import *
#from modeller import soap_protein_od
env = environ()
a = automodel(env, alnfile='2019S-6CRVA.ali',
knowns='6CRVA', sequence='2019S',
assess_methods=(assess.DOPE,
#soap_protein_od.Scorer(),
assess.GA341))
a.starting_model = 1
a.ending_model = 5
a.make()
from modeller import *
from modeller.scripts import complete_pdb
log.verbose() # request verbose output
env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology
env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# read model file
mdl = complete_pdb(env, '2019S.B99990002.pdb')
# Assess with DOPE:
s = selection(mdl) # all atom selection
s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='2019S.profile',
normalize_profile=True, smoothing_window=15)
import pylab
import modeller
def get_profile(profile_file, seq):
"""Read `profile_file` into a Python array, and add gaps corresponding to
the alignment sequence `seq`."""
# Read all non-comment and non-blank lines from the file:
f = file(profile_file)
vals = []
for line in f:
if not line.startswith('#') and len(line) > 10:
spl = line.split()
vals.append(float(spl[-1]))
# Insert gaps into the profile corresponding to those in seq:
for n, res in enumerate(seq.residues):
for gap in range(res.get_leading_gaps()):
vals.insert(n, None)
# Add a gap at position '0', so that we effectively count from 1:
vals.insert(0, None)
return vals
e = modeller.environ()
a = modeller.alignment(e, file='2019S-6CRVA.ali')
template = get_profile('6CRVA.profile', a['6CRVA'])
model = get_profile('2019S.profile', a['2019S'])
# Plot the template and model profiles in the same plot for comparison:
pylab.figure(1, figsize=(10,6))
pylab.xlabel('Alignment position')
pylab.ylabel('DOPE per-residue score')
pylab.plot(model, color='red', linewidth=2, label='Model')
pylab.plot(template, color='green', linewidth=2, label='Template')
pylab.legend()
pylab.savefig('dope_profile.png', dpi=65)